Bioinformatics training: transcriptomics
Protocol
We will follow the protocol described in Tophat2 bioinformatic protocol published in Nature protocol, 2012 for this course.
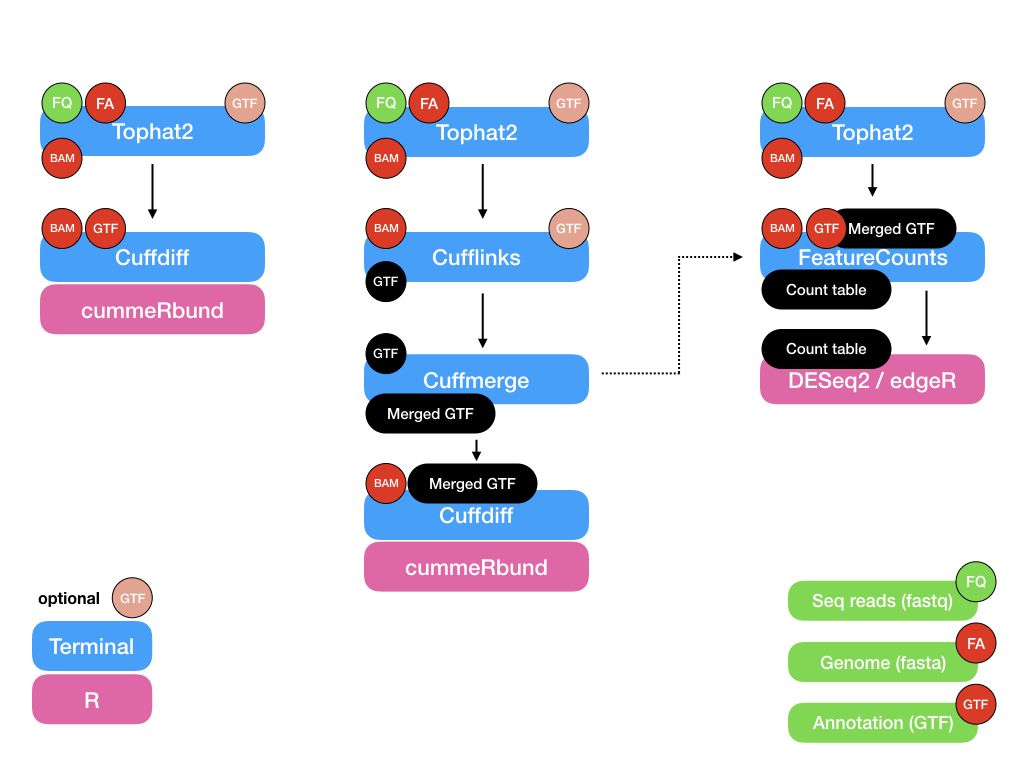
For simulated dataset:
- Tophat2 —> cufflinks —> cuffmerge —> cuffdiff —> cummeRbund
- Tophat2 —> cuffdiff —> cummeRbund
- Tophat2 —> featureCounts —> DESeq2/edgeR
For real life dataset
(Just because HISAT2 is new and much faster than Tophat2):
- HISAT2 —> StringTie —> Ballgown (There is an option to follow something similiar to cuffmerge as well)
- HISAT2 —> cufflinks pipeline (with or without cuffmerge) —> cummeRbund
- HISAT2 —> featureCounts —> DESeq2/edgeR
- HISAT2 worklow pipeline Nature protocol publication
Day 1
- Introduction of transcriptomic analyses
- Discusssion regarding reference-based and de novo approaches
- How to use tophat v2 - based on the Nature protocol (2012) paper
-
Download genome reference and create an index using bowtie2
- in /work/projects/nn9305k/home/
/transcriptome/ref $ ln ../../../../bioinf_course/transcriptomics/ref/Dm.BDGP6.dna.toplevel.fa . $ ls ../../../../bioinf_course/transcriptomics/ref/Dm.BDGP6.91.gtf . $ bowtie2-build Dm.BDGP6.dna.toplevel.fa Dm_BDGP6_genome - USAGE
$ bowtie2-build <Reference_fasta_file_name> <bowtie2-build_ref_index_name_that_you_will_use_later>
To do
- Align the given reads to the Drosophila genome using tophat v2
- create a new folder called tophat in /work/projects/nn9305k/home/
/transcriptome/ - create a slurm script with time=12:00:00, ntasks=16 and mem-per-cpu=12Gb
$ tophat -p 16 -G ../ref/Dm.BDGP6.91.gtf -o <tophat_output_folder_name> ../ref/Dm_BDGP6_genome <read1> <read2>
Day 2
- Discuss results from tophat v2 alignemnt
- Read about cufflinks and differential expression analysis pipeline
To do
- Run cufflinks on the tophat output
- Within tophat folder create a slurm script with time=12:00:00, ntasks=16 and mem-per-cpu=12Gb
$ cufflinks -p 16 -o <cufflinks_output_folder_name> <tophat_output_folder_name>/accepted_hits.bam
Day 3
- Discuss about (long) using tophat -> cufflinks -> cuffmerge -> cuffdiff pipeline (To find novel transcripts and genes)
- Discuss about (short) using tophat -> cuffdiff pipeline (To calculate differential expression for only known genes and transcripts)
To do
- Run cuffmerge and cuffdiff on Day 2’s cufflinks output
- create a text file and call it assemblies.txt and it should contain the information below
$ cat assemblies.txt
<cufflinks_output_folder_name_for_Con1_Rep1>/transcripts.gtf
<cufflinks_output_folder_name_for_Con1_Rep2>/transcripts.gtf
<cufflinks_output_folder_name_for_Con1_Rep3>/transcripts.gtf
<cufflinks_output_folder_name_for_Con2_Rep1>/transcripts.gtf
<cufflinks_output_folder_name_for_Con2_Rep2>/transcripts.gtf
<cufflinks_output_folder_name_for_Con2_Rep3>/transcripts.gtf
- Within tophat folder create a slurm script with time=12:00:00, ntasks=8 and mem-per-cpu=12Gb
$ cuffmerge -o <cuffmerge_output_folder_name> -g ../ref/Dm.BDGP6.91.gtf -s ../ref/Dm.BDGP6.dna.toplevel.fa -p 8 assemblies.txt - For the long pipeline, create a slurm script
cuffdiff -o cuffdiff_long_output -p 16 -L Con1_l,Con2_l <cuffmerge_output_folder_name>/merged.gtf Con1_Rep1_tophat/accepted_hits.bam,Con1_Rep2_tophat/accepted_hits.bam,Con1_Rep3_tophat/accepted_hits.bam Con2_Rep1_tophat/accepted_hits.bam,Con2_Rep2_tophat/accepted_hits.bam,Con2_Rep3_tophat/accepted_hits.bam - For the short pipeline, create a slurm script
cuffdiff -o cuffdiff_short_output -p 16 -L Con1_s,Con2_s ../ref/Dm.BDGP6.91.gtf Con1_Rep1_tophat/accepted_hits.bam,Con1_Rep2_tophat/accepted_hits.bam,Con1_Rep3_tophat/accepted_hits.bam Con2_Rep1_tophat/accepted_hits.bam,Con2_Rep2_tophat/accepted_hits.bam,Con2_Rep3_tophat/accepted_hits.bam - Check the above two scripts and identify the difference
Day 4
- Load cuffdiff output from short and long pipeline in R using cummeRbund
To do
- Link to cummeRbund manual: Manual
Day 5
- Introduction to counting reads
To do
- Link to featureCounts manual: Manual
Day 6-7
- DESeq2 and cummeRbund
To do
- Link to DESeq2 webpage: Link
- Link to DESeq2 manual: Link
- Link to cummeRbund webpage: Link
-
Link to cummeRbund manual: Link
- Link to DESeq2 Rscript: DESeq2.R
-
Link to cummeRbund Rscript: cummeRbund.R
- Link to video on FPKM, RPKM and TPM): Youtube video
- Link to video on DESeq2 Normalisation: Youtube video
Day 8
- Gene set enrichment analysis
- Gene Ontology
-
Pathway analyses - KEGG
- Gorilla
- g:Profiler
- Amigo
- DAVID
- PANTHER
Also, one can get the ortholog information from ENSEMBL
Day 9
- De novo assembly using Trinity
- Link to slide: presentation slides
- Trinity website: Github wiki
- Trinity worklow pipeline: Nature protocol publication