3 Input and usage
4 Usage
5 How to run
Bellow some short introduction on how to run the different tracks of the pipeline.
5.1 with containers
It is currently only possible to use the pipeline with containers. Tested using apptainer.
5.2 with conda
NOT IMPLEMENTED YET
5.3 SAGA users only
Please use apptainer as container. See here for configuration in SAGA (for all nextflow pipelines).
5.4 Which options can I choose
For an updated list of available options, please have a look at diplotopia/nextflow.config file (or online HERE). The nextflow.config file provides the options (and ssociated description) that are currently implemented in the pipeline. It also allows you to check what default options are used in the pipeline.
You do not need to change this file to change options that are used differently from run to run. The desired options can be given when launching the pipeline (see below) thanks to the priority order during nextlow configuration at runtime. However, the priority order means that you should put the options that you want to change after having mentionned any other configuration file that contains parameters affecting the pipeline options.
See nextflow documentation on configuration priority for more information.
Note: Available options will also be described for each track of the pipeline.
5.5 Running each track
General nextflow synthax
<path>/nextflow run <path>/main.nf -c <path/conf>/<selected>.config <optional: -c otherconfig> --outdir <path> --workdir <path> \
--track <track> --input <input.csv> --<option value> -profile <profile> Note: It is usually easier to use variables to put the full paths - the command gets clearer
For saga users:
The nextflow.config file and slurm configuration file saga_sapro.config are in a specific SAGA subdirectory in the pipeline. We need to use those configs for the pipeline to launch correctly.
# path of the pipeline directory
DIPLO="/cluster/projects/nn9305k/active/evezeyl/projects/Saprolegnia/git/diplotopia"
module purge
module load Java/17.0.4
MAIN="${DIPLO}/main.nf"
NF="/cluster/projects/nn9305k/bin/nextflow_23.04.4"
NFCONFIG="${DIPLO}/SAGA/nextflow.config"
CONFIG="${DIPLO}/SAGA/saga_sapro.config"
$NF run $MAIN -c $NFCONFIG -c $CONFIG --out_dir <path> -work-dir $USERWORK/diplotopia/<track> --track <track> --input <input.csv> --<option value> -profile apptainer -resume 2>&1 | tee <date>_nf.runlogFor the time being, I strongly recomend to add -resume and the external redirection to the log - it will be easier to debug (expected some problems at the beginning)
5.5.1 1. PURELY_RAW (under dev)
Pre-assembly. Detection and filtering out (optional) of contaminated reads, prior to assembly.
Options implemented:
5.5.2 2. TRYASSEMBLY (under dev)
- multiple assemblies test
Options implemented:
5.5.3 3. FILTER_CONTIGS
Purpose: Filtering out contigs that do not represent the genome of the organism of interest
5.5.3.1 input preparation
5.5.3.2 Options curently implemented:
blastDB = nullplease provide the path of your blast database (once at installation)ranked_taxo_file = null” please provide the path of NCBI taxonomic file: “rankedlineage.dmp” (once at installation)positive_filter = nullrlang filtering expression, any contigs detected as corresponding (positive) to this filtering criteria will be kept in the dataset. Example “phylum == ‘Oomycota’” . ! OOPS : Be aware that the external brackets need to be double. See here for additional examples.evalue_min = 1e-20query results where positive filtering expression is TRUE, are only considered as valid if the blast \(evalue \geq 1e-20\)perc_identity_min = 95query results where positive filtering expression is TRUE, are only considered as valid if the blast identify measure of the query \(\geq 95\%\) identity with the target sequence.
5.5.3.3 Example of command for SAGA users:
5.5.4 4. COMPASS
Purpose: Evaluation of assembly quality and compleness, for comparison of assemblies with different methods/assemblers. This in order to guide the choice of which assembly to use for downstream analyses.
5.5.4.1 input preparation
“input.csv” : A comma delimited file containing the following fields: ID,assembly,longreads,R1,R2,type,comments
Note: The field comments is not used during analysis but must be present and can be left empty. Its purpose to allow you to put some notes in your input files, as long as you do not use commas as separator. You can eg. write several comments using | instead of , to write long comments.
If you only have long reads :
- Set
NAin “R1” and “R2” fields - Set
longin “type” fields
If you only have short reads :
- Set
NAin “longreads” field - Set
shortin “type” field
If you have both long and short reads :
- Set
hybridin “type” field
5.5.4.2 Options curently implemented:
Contigs classification and removal of contaminated contigs - remove_contaminants = "no" if “yes” then activates FILTER_CONTIGS with the selected parameters
Detection of contaminants in assemblies - krakenDB = null please provide the path of the kraken database to use (eg. once at installation), see also how to install. Option for detecting contaminants in assemblies
Assembly quality assessment with QUAST - which_quast = "ref" : options: “ref”, “assembly”, “both” : if quast is to be run with an external reference and/or using an internal assembly was reference.
Path of the external genome/asssembly and of its annotations (.gff), to be use as external reference for assembly quality assessment using quast. (if option)
Can eg. be provided at laucn time using --quast_ref <path> - quast_ref = null
- quast_annot = null
Path of the internal assembly that is used by QUAST - quast_ref_assembly = null
QUAST native options: - min_contig = 500 only shows contigs of size $ ; bp$ - plot_format = "svg"
Assembly completeness assessment with BUSCO
lineage_dataset = nullA list of lineages to use, separated by commas. Ex.: “alveolata_odb10,stramenopiles_odb10,eukaryota_odb10”busco_download_path = nullplease provide the path where busco lineages databases have been donwloaded (should be ending withbusco_downloads)
| #### Example of command for SAGA users: |
| ```bash DIPLO=“/cluster/projects/nn9305k/active/evezeyl/projects/Saprolegnia/git/diplotopia” |
| module purge module load Java/17.0.4 |
| MAIN=“\({DIPLO}/main.nf" NF="/cluster/projects/nn9305k/bin/nextflow_23.04.4" NFCONFIG="\){DIPLO}/SAGA/nextflow.config” CONFIG=“${DIPLO}/SAGA/saga_sapro.config” |
| # Synthax $NF run $MAIN -c $NFCONIG-c \(CONFIG --outdir <path> -work-dir <\)USERWORK/diplotopia/COMPASS –track coverage –input input.csv [– |
if you are relaunching add -resume |
All the optional parameters are given in nextflow.config file. To change those, use : --<parameter_name> <value> |
| ### 5. HAPLOPURGE |
| Purpose: Obtaining an haploid representation of the genome (HaploSSembly) |
This track takes advantage of the option -resume implemented in nextflow to run a pipeline that would otherwise require decision/user input; which is otherwise in opposition to the principe of pipeline running. |
| Running this track step by step with the -resume option will allow you to find the parameters that should be used, and allow you to rerun all the steps at once when you know what parameters to use, eg. prior to writing your definitive results. |
5.5.4.3 input preparation
“input.csv” : A comma delimited file containing the following fields: ID,assembly,longreads,R1,R2,type,haplo_low,haplo_mid,haplo_high,comments
It is an extension of the [COMPASS Track] input, as it includes additional fields haplo_low, haplo_mid, haplo_high which are required for purging haplotigs.
If you do not know which parameters to employ in haplo_low, haplo_mid, haplo_high fields yet, please Set NA in those fields.
haplo_low, haplo_mid, haplo_high corresponds to purge haplotigs low, mid, high parameters (step1) and need to be provided in input.csv. This allows you to check different assemblies with data (leading to different reads coverage).
5.5.4.4 Options curently implemented:
Running the track step by step:
--purge_phase = null: indicate either “hist” (1) OR “cov” (2) OR “purge” (3) OR “all”
This parameters allows you to make the pipeline stop when you need to take a decision and adjust the parameters of the pipeline step by step. First using : hist, then cov, followed by purge until you are satistified with the results. all will run everything, allowing you to get a clean run with all the parameters set (eg. prior to writing an article).
The results of histstage will allow you to choose the haplo_low, haplo_mid, haplo_high parameters, which are the coverage cutoff parameters “low, mid, and high”, defined by purge haplotigs software
 > Citat: Purge Haplotigs sofware https://bitbucket.org/mroachawri/purge_haplotigs/src/master/
> Citat: Purge Haplotigs sofware https://bitbucket.org/mroachawri/purge_haplotigs/src/master/
The improvement of the assembly, towards an HaploSSembly is then under control of the following parameters:
purgehaplotigs_maxdepth = 200: purge haplotigsdepthparameter (step1). The maximum cutoff for depth. Adjust higher than your expected coverage, and to obtain a readeable histogram.haplo_junk = 80: purge haplotigsjunkparameter (step2). Junk coverage threshold for haplotigs (integer). % contig where low/high, if \(\geq\) % then is assigned as junk. Junk is not removed if > 100haplo_suspect = 80: purge haplotigssuspectparameter (step2). Coverage threshold for suspect haplotigs (integer). Contig is suspect if diploid coverage is \(leq\) % of the contig > aka. alleles at a locus are given in a different contig.haplo_align_cov = 70: purge haplotigsalign_covparameter (step3). The read covearge cutoff at which the contig is estimated to be haploid. Bellow this percentage, the contig is considered as an haplotig.
haplo_max_match = 250: purge haplotigsmax_matchparameter (step3). The % read covearge cutoff at which the contig is estimated to be repetitive Above this percentage, the contig is considered to be repeated.
5.5.4.5 Example of command for SAGA users:
Example of initial run
DIPLO="/cluster/projects/nn9305k/active/evezeyl/projects/Saprolegnia/git/diplotopia"
module purge
module load Java/17.0.4
MAIN="${DIPLO}/main.nf"
NF="/cluster/projects/nn9305k/bin/nextflow_23.04.4"
NFCONFIG="${DIPLO}/SAGA/nextflow.config"
CONFIG="${DIPLO}/SAGA/saga_sapro.config"
# first time
$NF run $MAIN -c $NFCONIG -c $CONFIG --outdir <path> -work-dir <$USERWORK/diplotopia/HAPLOPURGE --track haplopurge --input input.csv [--<optional params> ] --purge_phase hist -profile apptainer -resume 2>&1 | tee haplopurge_hist.runlog
# rerun all (do not resume)
$NF run $MAIN -c $NFCONIG -c $CONFIG --out_dir . -work-dir -work-dir <$USERWORK/diplotopia/HAPLOPURGE --track haplopurge --input input.csv --haplo_junk 105 -profile apptainer --purge_phase all | tee haplopurge_all.runlog5.5.5 6. VARWRRUM:
Purpose: Variant calling (incl. at the population level, filtering and normalisation)
Current implementation: Variants are called using short reads (illumina)
5.5.5.1 input preparation
You will need to prepare two different input files for this track, both comma delimited files (.csv)
input.csv : A comma delimited file, where you will indicate the information about samples you want to call variants for. The file must contain the following fields: ID,R1,R2,ploidy,comments .
ref_input.csv : A comma delimited file, where the information conserning the haploid reference that will be used for variant calling on all the samples is indicated. The file must contain the following fields: ID,R1,R2,ploidy,comments
R1 and R2 fields are the path of respective reads corresponding to each sample (or to the haploid reference). Note: Short reads can be used for softmasking regions with high coverage in the reference. ! Not implemented yet
ID is the name for either the samples of the reference.
The field comments is not used during analysis but must be present and can be left empty. Its purpose to allow you to put some notes in your input files, as long as you do not use commas as separator. You can eg. write several comments using | instead of , to write long comments.
5.5.5.2 Options curently implemented:
Running the track step by step:
Allows parameter adjustments, see eg. masking the reference, filtering mapped reads and filtering variants parameters.
phase = null: a choice to enter “prep_ref” (1) OR “prep_samples” (2) OR “call_variants” (3) This parameters allows you to run the pipeline step by step, when used in conjunction with-resume. Notecall_variantswill run everything if this has not been done before. This allows you to get a clean run with all the parameters set approriately (eg. prior to writing an article).
Trimming reads prior to mapping: Trim Galore parameters - phred_score = 15 - error_rate = 0.1 - minlength = 20
Soft masking of the reference: bbmask parameters - bbmask_entropy = 0.7 bbmask entropy parameter
bbmask_maxcov = nullbbmask maxcov parameter (optional). ! Currently not implemented
Mapping reads: BWA mem parameters - bwamem_args = "-a -M" a flexible argument : parameters to pass to bwa mem.
Default parameters:
kmer size can be adapted by adding -k eg. “-k 19”
Filtering of mapped reads: Samtools parameters
min_mapq = 30reads with MAPQ (mapping quality) \(/leg 30\) are filtered out. (MAPQ given as a phread value)filterout_tag = "-F 0x0008 -F 0x0100 -F 0x0400 -F 0x0800 -F 0x0004"reads from which those tags are present are filtered out. See sammtools flags for flags code.
Note: This will filter out reads where one is unmapped/unpaired, and will filterout positions that are secondary alignment and supplementary alignments.
Variant Calling parameters: Freebayes
vcf_ref = null(optional) a vcf file that is used as reference to force calling variants at particular alleles, eg. to standardize with a previous population analysis. Note that the variant calling MUST have used the same assembly/haploSSembly as reference for variant calling.chunk_size = 100000chunks the reference to parallelize variant calling to use Freebayes parallelfreebayes_args = "--min-coverage 10"flexible argument : parameters to pass to freebayes to control variant calling. By default the only option is to not call variants from which reads coverage (after having filtered the mapped reads) are represented by in less than 10 reads.
Freebayes default parameters to call variants for diploid organisms: --min-alternate-count 2 --min-alternate-fraction 0.2 have not been modified and are thus employed here; see Freebayes instructions
Note: The list of available options can be optained with freebayes by typing
freebayes -h. For convenience it has been copied and linked [here][3]
Variants Filtering parameters: bcftools - filter_args = "-i 'QUAL>=100 && FMT/DP >= 30 && FMT/QA >= 100'" flexible argument : parameters to pass to freebayes to control the filtering of valid variants.
Par default \(QUAL \geq 100\) means that we require variants to be called with a minimum quality/error rate corresponding to phred 100. There is a very high probability that there is a polymorphism at this loci 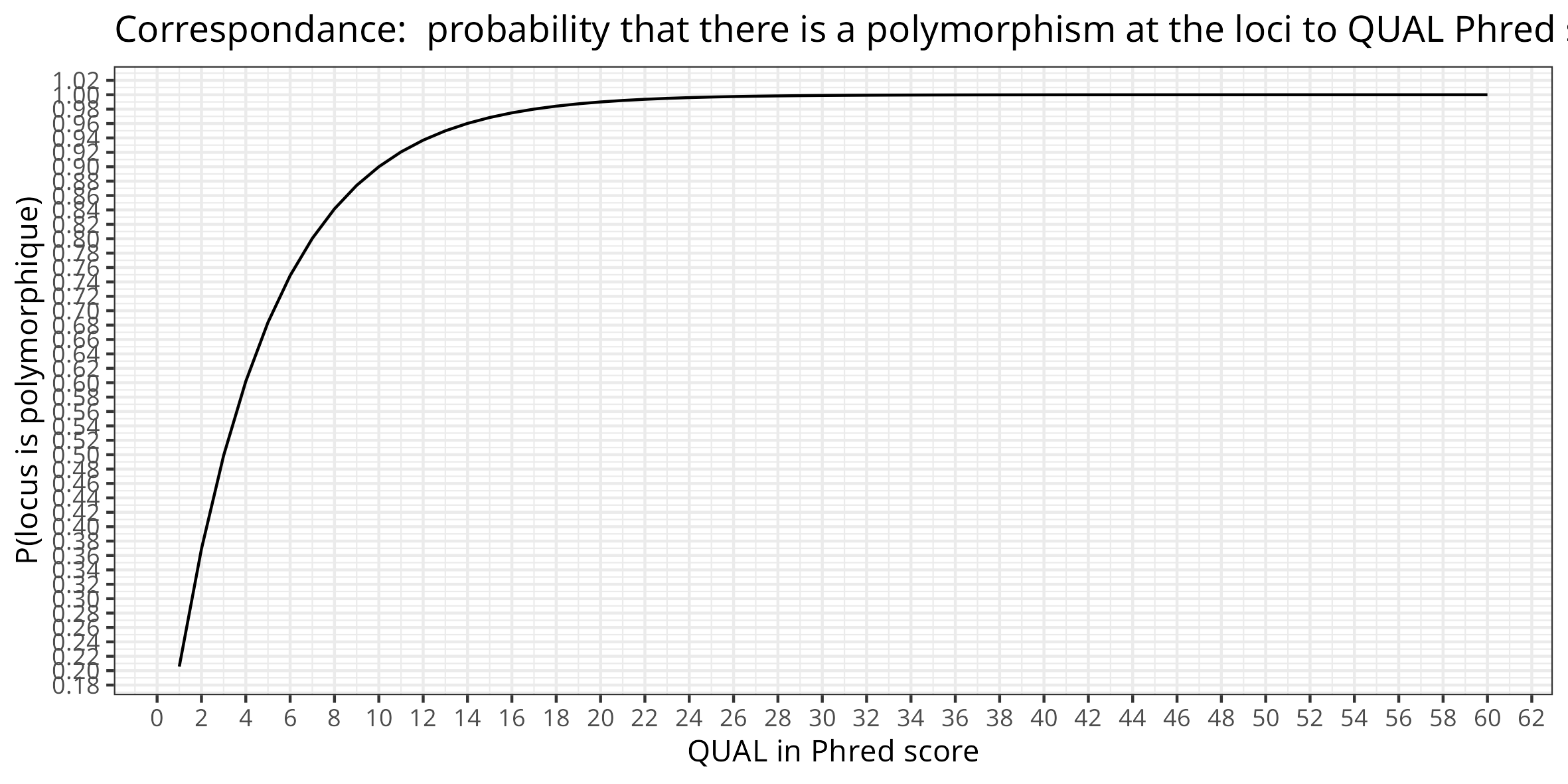
Par default \(FMT/DP \geq 30\) means that variants are not called if the Read Depth at the loci is below 30.
Par default \(FMT/QA \geq 100\) sum of quality of the alternate observations (phred) is required for a variants to be called at specific site.
Note: The headers/fields that are reported by freebayes have been copied and linked [here][4] and can be employed to adjust the filtering parameters according to your requirements.
Other
Please review purgehaplotigs_maxdepth parameter for completness at this parameter is used to re-estimate the histograms of ploidy with purge haplotigs (informational)
5.5.5.3 Example of command for SAGA users:
DIPLO="/cluster/projects/nn9305k/active/evezeyl/projects/Saprolegnia/git/diplotopia"
module purge
module load Java/17.0.4
MAIN="${DIPLO}/main.nf"
NF="/cluster/projects/nn9305k/bin/nextflow_23.04.4"
NFCONFIG="${DIPLO}/SAGA/nextflow.config"
CONFIG="${DIPLO}/SAGA/saga_sapro.config"
REFCSV="<path>/ref_input.csv"
INPUTCSV="<path>/input.csv"
$NF run $MAIN -c $NFCONIG -c $CONFIG --out_dir . -work-dir $USERWORK/diplotopia/VARWRRUM \
--track varwrrum --input $INPUTCSV --ref_input $REFCSV --phase call_variants \
-profile apptainer -resume 2>&1 | tee varwrrum_nf.runlog5.5.5.4 Additional information
You might want to refere to this document if you wish to adjust filtering of mapped reads SAM/BAM format specification v1
You might want to refere to this document if you wish to adjust calling and filtering of variants: The variant Call Format v4.2
5.6
6 Parameters description
Note: A memo to recognize the type of parameters when running the pipeline
Nextflow parameters :
-nextflow running options (eg. profiles and configuration files)pipeline parameters :
--options for running the different tools of the pipeline
**Input and output**
--input: Input csv file
--out_dir: Output directory name
Workflow-specific parameters
--
**General parameters**
**Time-related parameters**
--time_multiplier: Default value 1. If increased to 2, doubles the time requested by the system for